





¿Qué es la enfermedad de Gaucher?
La enfermedad de Gaucher es la Enfermedad de Deposito Lisosomal, EDL, más común. Existen 3 tipos, en función de la afectación neurológica¹. El 90% de los pacientes padecen la no neuropática, es decir, la enfermedad de Gaucher tipo 1 (EG1)². Afecta por igual a hombres y mujeres³.
Su inicio y gravedad, así como su presentación clínica inicial, varía considerablemente entre pacientes, resultando un reto para el diagnóstico²: el tiempo medio desde la primera aparición de los síntomas de la enfermedad hasta el diagnóstico es de aproximadamente 4 años, pudiéndose retrasar en ocasiones hasta 10 años⁴.
Es un trastorno autosómico recesivo caracterizado por la deficiencia de la enzima glucocerebrosidasa (GCB) debido a mutaciones en el gen GBA1, lo cual conduce a su vez a la acumulación de un glucocerebrósido anormal (glucosilceramida) en los lisosomas, desencadenando finalmente un amplio espectro de manifestaciones fenotípicas⁵.
En el siguiente vídeo, la Dra. Pilar Giraldo nos cuenta qué es la Enfermedad de Gaucher y cómo impacta en la calidad de vida de los pacientes.

Clasificación de la enfermedad de Gaucher⁶
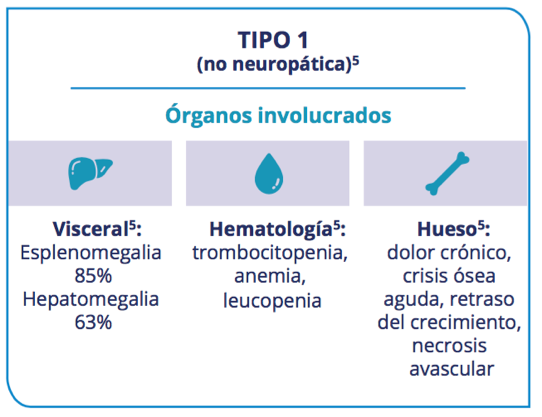
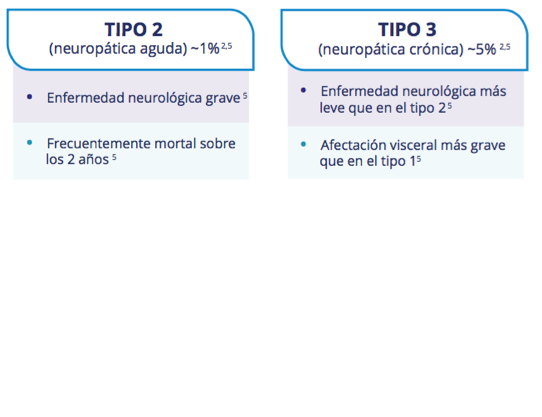
El dolor óseo y la fatiga crónica son los síntomas más debilitantes de la EG, pudiendo interferir con el colegio, trabajo o las actividades sociales10
Signos y síntomas
Entre los síntomas comunes de la EG se incluyen⁶:
- Esplenomegalia
- Afectaciones óseas como dolor, necrosis avascular, fracturas
- Hepatomegalia
- Anemia / Trombocitopenia
- Hiperferritinemia
- Gammapatía
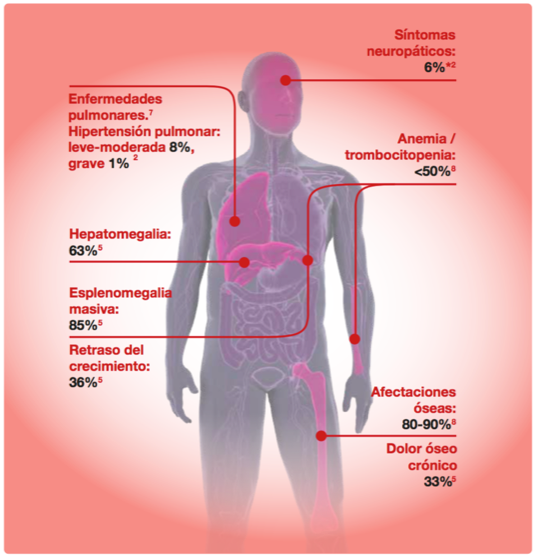
Los signos y síntomas más comunes en niños observados en el momento del diagnóstico fueron: esplenomegalia (95%), hepatomegalia (87%) y enfermedad ósea radiológica (81%), trombocitopenia (50%), anemia (40%), retraso del crecimiento (34%), dolor óseo (27%) y crisis ósea (9%)¹¹.
No obstante, aunque algunos pacientes con EG presentan síntomas desde la niñez hasta la edad adulta, otros permanecen asintomáticos³.
Los niños asintomáticos con diagnóstico de EG cuyos hermanos están afectados, deben ser monitorizados al menos cada 6 meses. Éstos pueden presentar un curso similar de la enfermedad, y requieren monitorización adicional¹¹.
El reconocimiento temprano de la EG por parte de los pediatras es importante ya que la intervención temprana puede disminuir la morbilidad y section-bg conoceucir el riesgo de complicaciones posteriores¹².
Diagnóstico de la Enfermedad de Gaucher
La EG se diagnostica comúnmente en la infancia o la adolescencia, aunque la edad media en el diagnóstico de la EG1 es de 30 a 40 años¹³.
Un retraso prolongado en el diagnóstico de la EG puede resultar en problemas clínicos irreversibles¹⁴.
Los pacientes con EG pasan por una media de tres especialistas antes de ser diagnosticados. De estos diagnósticos, un 75% es realizado por hematólogos15.
Los primeros indicios de la EG pueden verse mediante la aparición de células de Gaucher en biopsias de médula ósea o hígado¹.

El diagnóstico específico de la EG se realiza mediante1,6:
- Prueba de gota de sangre seca: ofrece un método de colección de sangre para realizar el análisis de glucocerebrosidasa ácida en leucocitos.
- Combinación de la prueba anterior con la identificación de mutaciones en el gen que codifica GBA1: Se recomienda la secuenciación completa de este gen para establecer el diagnóstico molecular y la mejor detección de portadores.

PRUEBA DE GOTA DE SANGRE SECA
Se considera adecuado informar y ofrecer la posibilidad de realizar pruebas diagnósticas a los familiares asintomáticos en riesgo, de modo que aquellos con deficiencia de la enzima glucocerebrosidasa puedan beneficiarse del diagnóstico y tratamiento tempranos en caso de ser necesario16.
Un diagnóstico temprano es esencial para evitar complicaciones de la enfermedad17.

DIAGNÓSTICO DE LA EG1 EN PEDIATRÍA Y EN LA EDAD ADULTA
En el siguiente vídeo, la Dra. del Toro (Hospital Universitari Vall d’Hebron, Barcelona) y la Dra. Morales (Hospital Universitario 12 de octubre, Madrid), cuentan las claves del diagnóstico de la EG1 en pediatría y en edad adulta. Accede para descubrir cuáles son los síntomas que pueden indicar la existencia de una EG1 en pediatría o porqué es necesario un diagnóstico precoz.
VIVIR CON LA ENFERMEDAD DE GAUCHER
Una vez diagnosticado con EG, cada paciente, cuya esperanza de vida suele estar entre los 60-90 años18 deberá evaluarse a nivel individual. Su enfermedad se clasificará según un sistema de puntuación de la intensidad clínica19.
El objetivo de la evaluación es controlar los signos, síntomas y complicaciones asociadas a la EG que puedan ser indicativos de la necesidad de tratamiento, como anemia, trombocitopenia, hemorragia, hepatoesplenomegalia, manifestaciones esqueléticas y anomalías de la función hepática o pulmonar20.
El objetivo fundamental de la terapia de la EG es evitar que la enfermedad progrese con complicaciones graves. Esto se puede conseguir mediante: terapia de sustitución enzimática (TSE) o terapia de reducción de sustrato (TRS)5.
Además, también existen ciertos tratamientos sintomáticos para aliviar los síntomas los cuales deben ser administrados por un equipo multidisciplinar con experiencia en esta enfermedad16.
¿EN QUÉ PODEMOS AYUDARTE?
Acrónimos:
EDL: Enfermedades de Depósito Lisosomal
EG: Enfermedad de Gaucher;
EG1: Enfermedad de Gaucher tipo 1
GCB: Glucocerebrosidasa
GBA1: Gen codificante de la enzima glucocerebrosidasa
TSE: Terapia de Sustitución Enzimática
TRS: Terapia de Reducción de Sustrato
Referencias:
- 1. Dandana A, Ben Khelifa S, Chahed H, Miled A, Ferchichi S. Gaucher Disease: Clinical, Biological and Therapeutic Aspects. Pathobiology. 2016;83(1):13–23.
- 2. Cassinerio E, Graziadei G, Poggiali E. Gaucher disease: A diagnostic challenge for internists. Eur J Intern Med. 2014; 25(2):117-24
- 3. Huang WJ, Zhang X, Chen WW. Gaucher disease: a lysosomal neurodegenerative disorder. Eur Rev Med Pharmacol Sci. 2015 Apr;19(7):1219-26.
- 4. Mistry PK, Sadan S, Yang R, Yee J, Yang M. Consequences of diagnostic delays in type 1 Gaucher disease: the need for greater awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. Am J Hematol. 2007;82(8):697-701
- 5. Nagral A. Gaucher disease. J Clin Exp Hepatol. 2014;4(1):37-50
- 6. Revel-Vilk S, Szer J, Mehta A, Zimran A. How we manage Gaucher Disease in the era of choices. Br J Haematol. 2018;182(4):467-480.
- 7. Pastores GM, Weinreb NJ, Aerts H, Andria G, Cox TM, Giralt M, Grabowski GA, Mistry PK, Tylki-Szymańska A. Therapeutic goals in the treatment of Gaucher disease. Semin Hematol. 2004;41(4 Suppl 5):4-14.
- 8. Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher's disease. Lancet. 2008;372(9645):1263-71
- 9. Weinreb NJ, Deegan P, Kacena KA, et al. Life expectancy in Gaucher disease type 1. Am J Hematol. 2008;83(12):896-900
- 10. Hayes RP, Grinzaid KA, Duffey EB, Elsas LJ 2nd. The impact of Gaucher disease and its treatment on quality of life. Qual Life Res. 1998 Aug;7(6):521-34
- 11. Kaplan P, Baris H, De Meirleir L, Di Rocco M, El-Beshlawy A, Huemer M, Martins AM, Nascu I, Rohrbach M, Steinbach L, Cohen IJ. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013; 172(4):447-58
- 12. Kaplan P, Andersson HC, Kacena KA, Yee JD. The clinical and demographic characteristics of nonneuronopathic Gaucher disease in 887 children at diagnosis. Arch Pediatr Adolesc Med. 2006;160(6):603-8.
- 13. Ferreira CR, Gahl WA. Lysosomal storage diseases. Transl Sci Rare Dis. 2017;2(1-2):1-71
- 14. Linari S, Castaman G. Clinical manifestations and management of Gaucher disease. Clin Cases Miner Bone Metab. 2015;12(2):157-64.
- 15. Thomas AS, Mehta AB, Hughes DA. Diagnosing Gaucher disease: an on-going need for increased awareness amongst haematologists. Blood Cells Mol Dis. 2013; 50(3):212-7.
- 16. Pastores GM, Hughes DA. Gaucher Disease. Junio 2018. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK1269/. Útimo acceso: febrero 2022.
- 17. Johnson BA, Dajnoki A, Bodamer O. Diagnosis of lysosomal storage disorders: Gaucher disease. Curr Protoc Hum Genet. 2014; 82:17.15.1-6.
- 18. Mehta A. Epidemiology and natural history of Gaucher's disease. Eur J Intern Med. 2006;17 Suppl:S2-5.
- 19. Harmanci O, Bayraktar Y. Gaucher disease: new developments in treatment and etiology. World J Gastroenterol. 2008; 14(25):3968-73.
- 20. Jmoudiak M, Futerman AH. Gaucher disease: pathological mechanisms and modern management. Br J Haematol. 2005; 129(2):178-88.
* Paciente ficticio. Imagen de libre uso
◊ Imagen de libre uso
